All content on this site is intended for healthcare professionals only. By acknowledging this message and accessing the information on this website you are confirming that you are a Healthcare Professional. If you are a patient or carer, please visit the Lymphoma Coalition.
The Lymphoma Hub uses cookies on this website. They help us give you the best online experience. By continuing to use our website without changing your cookie settings, you agree to our use of cookies in accordance with our updated Cookie Policy
Introducing

Now you can personalise
your Lymphoma Hub experience!
Bookmark content to read later
Select your specific areas of interest
View content recommended for you
Find out moreThe Lymphoma Hub website uses a third-party service provided by Google that dynamically translates web content. Translations are machine generated, so may not be an exact or complete translation, and the Lymphoma Hub cannot guarantee the accuracy of translated content. The Lymphoma Hub and its employees will not be liable for any direct, indirect, or consequential damages (even if foreseeable) resulting from use of the Google Translate feature. For further support with Google Translate, visit Google Translate Help.









iwCLL 2017 | On the Origin of CLL Evolution
Bookmark this article
The sixth session at this year’s iwCLL was titled “Genetic Changes Involved in the Progression and Evolution of CLL”, and was jointly chaired by Christopher Vakoc (Cold Spring Harbor Laboratory, Cold Spring Harbor, New York, New York) and Nicole Lamanna (New York Presbyterian and Columbia University Medical Center, New York, New York).
The first talk during this session was given by Dan Landau, MD, PhD, from Weill Cornell Medical College, New York, USA, and was titled “On the Origin of CLL Evolution.”
Landau began by stating that tumor evolution is a central obstacle to curative cancer therapy. If treatment is used on a cancer which displays intra-tumoral heterogeneity, then tumor evolution can result in progression, relapse, and treatment resistance. CLL is remains incurable despite very effective therapies. CLL allows studying evolution in its natural environment.
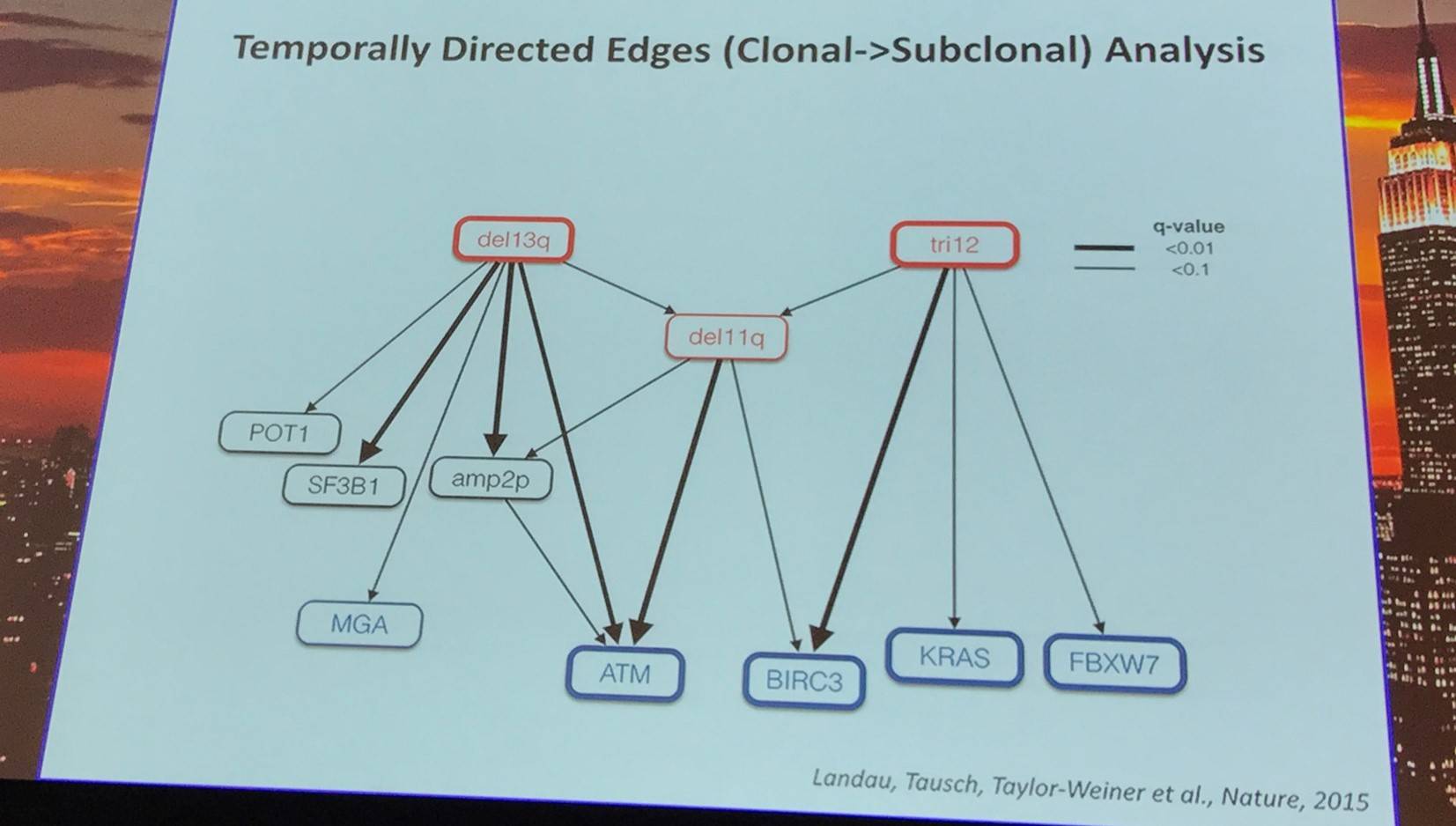
Landau et al. (Cell, 2013) studied intra-tumoral heterogeneity in 149 CLL cases by integrating Whole-Exome Sequence (WES) and copy number to measure the fraction of cancer cells harboring each somatic mutation. They identified driver mutations as predominantly clonal (such as MYD88, trisomy 12, and del(13q)) or sub-clonal (e.g. SF3B1 and TP53), corresponding to earlier and later events in CLL evolution.
In addition, they sampled leukemia cells from 18 patients at two different time points. Ten of twelve CLL cases treated with chemotherapy (but only one of six without treatment) underwent clonal evolution, predominantly involving subclones with driver mutations (e.g. SF3B1 and TP53) that expanded over time.
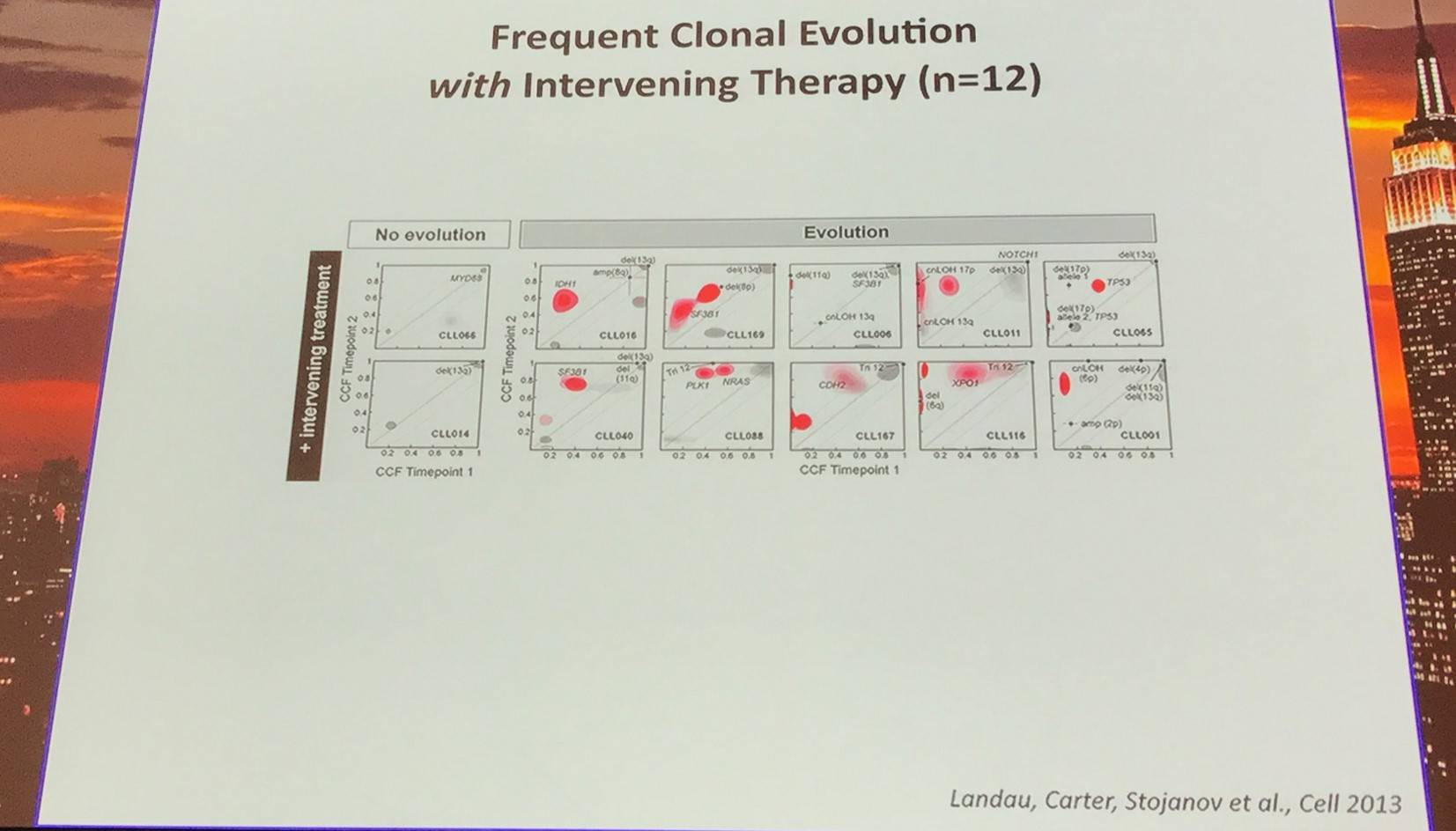
Red contour denote a CCF increase of >0.2
Furthermore, Landau et al. (Cell, 2013) found that the presence of a sub-clonal driver mutation was an independent risk factor for rapid disease progression.
In a later study, Landau et al. (Nature, 2015) investigated which genetic alterations drive tumorigenesis and how they evolve over the course of disease and therapy. They found that only 3% of CLLs are un-evolved at relapse and the remaining 97% of CLLs were significantly evolved in relapse. In 18 patients, evolution involved the expansion of a sub-clone already detected in the pre-treatment sample.
They identified 44 recurrently mutated genes and 11 recurrent somatic copy number variations through WES of 538 CLL and matched germline DNA samples, 278 of which were collected in a prospective clinical trial. These include previously unrecognized putative cancer drivers (RPS15, IKZF3) and collectively identify RNA processing and export, MYC activity, and MAPK signaling as central pathways involved in CLL.
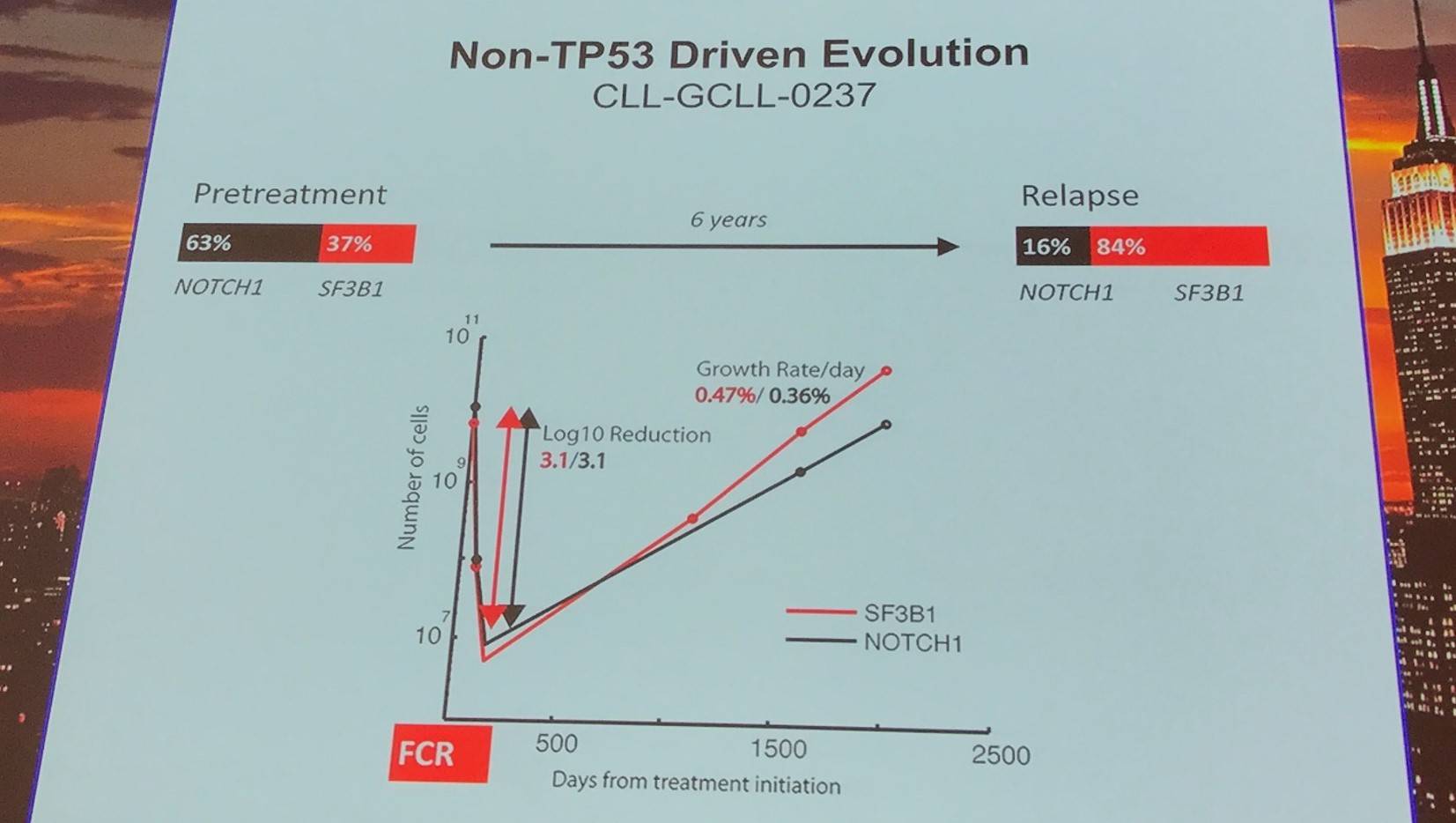
Non-TP53 driven evolution was found in 8 cases, relying more on competitive outgrowth and not selective resistance.
|
|
Pre-treatment |
Relapse |
p-value |
|---|---|---|---|
|
Average log10 reduction with therapy |
3.9 |
3.9 |
0.9 |
|
Average re-population rate (per day) |
0.5% |
0.5% |
0.03 |
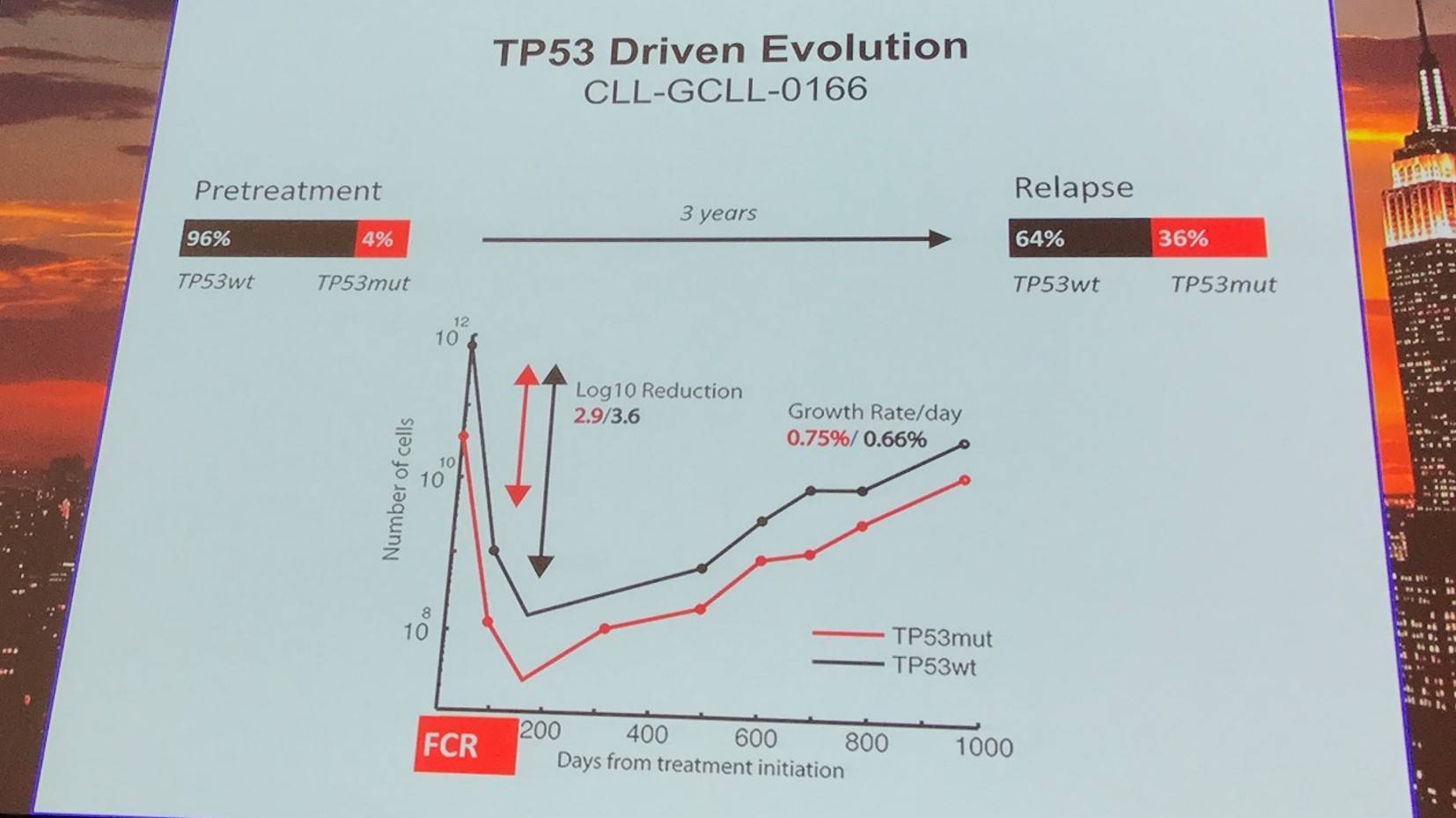
TP53 driven evolution was found in 10 cases, relying more on selective resistance compared to competitive outgrowth.
|
|
TP53 wild type |
TP53 mutant |
p-value |
|---|---|---|---|
|
Average log10 reduction with therapy |
3.8 |
32.4 |
0.02 |
|
Average re-population rate (per day) |
0.6% |
0.8% |
0.07 |
Moving on from this, Dan Landau also briefly discussed drivers of resistance to ibrutinib:
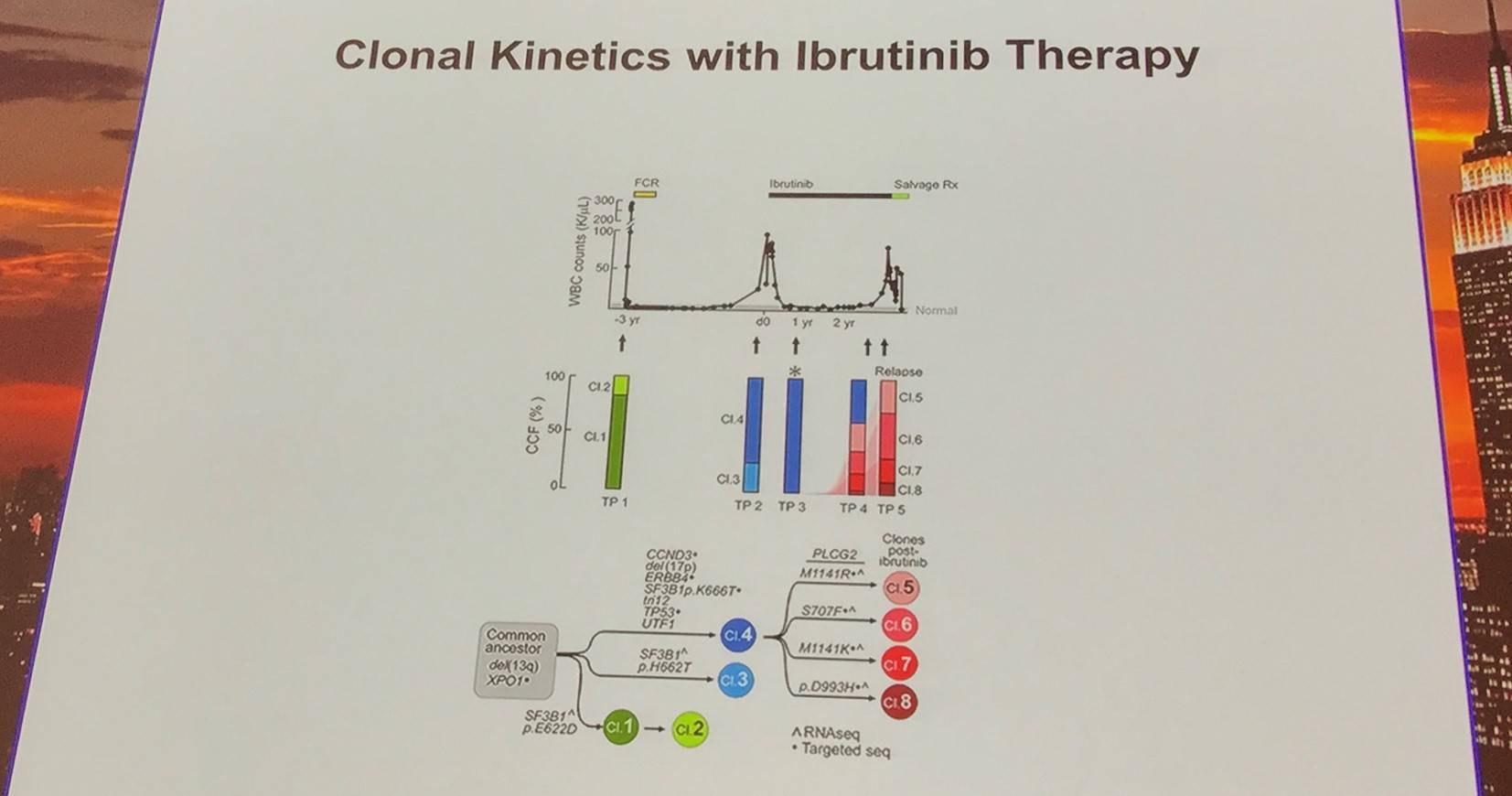
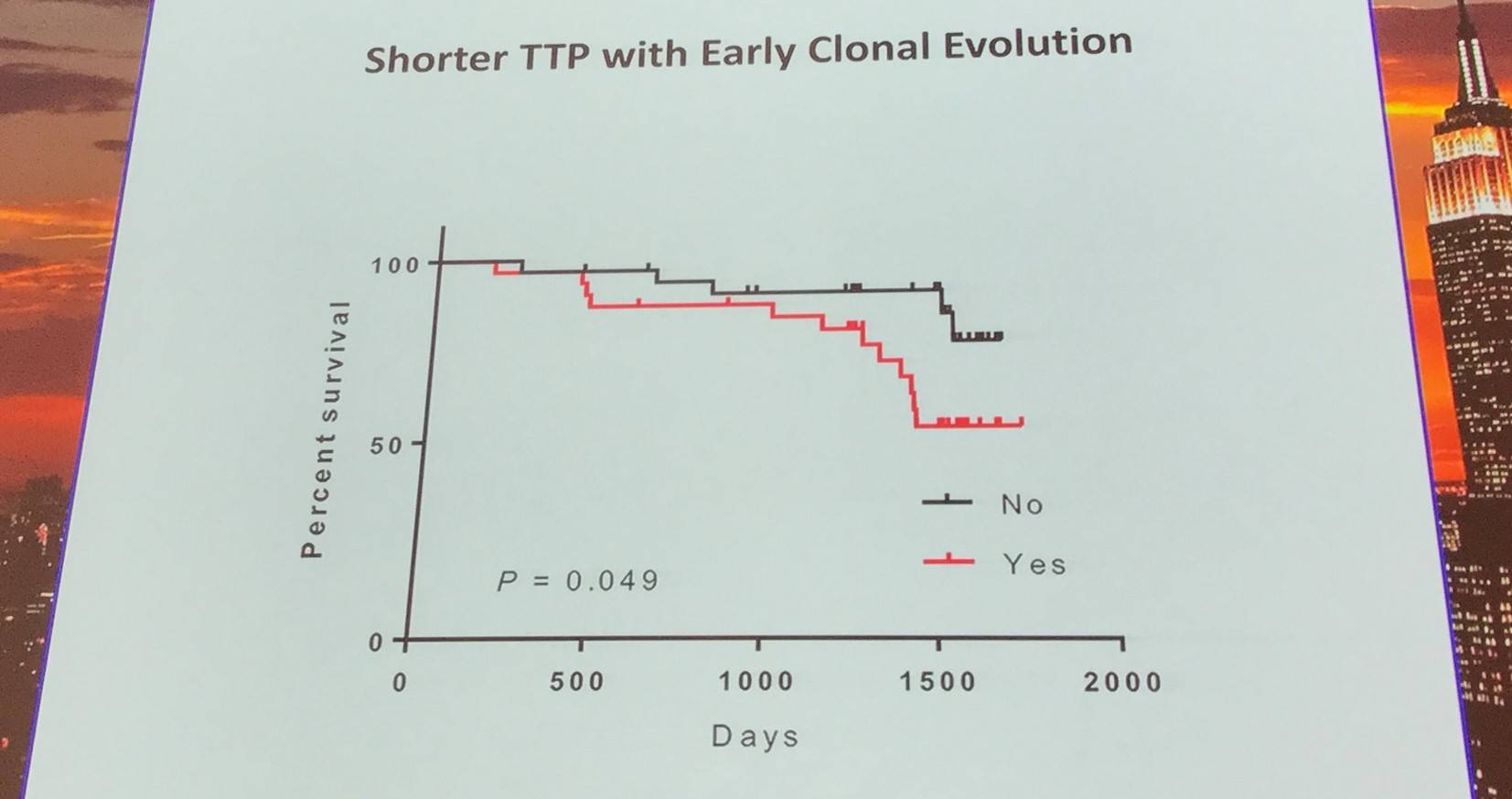
Additionally, the early evolutionary landscape of ibrutinib treated CLL was discussed. Serial WES of ibrutinib treated patients (n=61) was carried out. Cohort A (n=45) were treated with 420mg/d ibrutinib and cohort B (n=16) received 400mg/d. The median number of samples per patient was 3 (range, 3–5). Frequent clonal evolution was found early in therapy (30/61).
- Landau D. On the Origin of CLL Evolution. XVII International Workshop on Chronic Lymphocytic Leukemia; 2017 May 12–15; New York, USA.

Understanding your specialty helps us to deliver the most relevant and engaging content.
Please spare a moment to share yours.
Please select or type your specialty
 Thank you
Thank youRelated articles
Newsletter
Subscribe to get the best content related to lymphoma & CLL delivered to your inbox